
”hisat2“ 的搜索结果

基于图的基因组比对和HISAT2和HISAT基因型的基因分型 接触 ( )和 ( ) 抽象的 下一代测序技术的飞速发展极大地改变了我们进行基因组规模分析的能力。 用于大多数基因组分析的人类参考基因组仅代表少数个体,从而...
HISAT2Aligner HISAT2是一种快速灵敏的比对程序,可将下一代测序读数(DNA和RNA)映射到人类基因组(以及单个参考基因组)。 请参阅了解该算法的详细信息。
(51100+57804+33582+277922+1144382)/(4165142)=0.4803
原以为是数据出现了问题,经检查发现原来是hisat2指令输错了……把--rna 误写成了--tna……看到有其他朋友把‘-’打成‘--’也会出现一样的报错,总之写完指令还是要多检查一下。使用hisat2进行比对时出现如题报错。
Hisat2是一款短序列比对的工具,主要用于转录组数据的比对,是Hisat比对工具的升级版。Hisat2优化了索引建立的策略,采用了新的比对策略,使其与Bowtie/TopHat2等软件相比具有更高的敏感性和更快的运算速度。Hisat2...
p #多线程数 -x #参考基因组索引文件目录和前缀 -1 #双端测序中一端测序文件 -2 #同上 -S #输出的sam文件;...2、寻找新的可变剪切isoform,RNA的可变剪切,需要看外显子差异,用TopHat, HISAT2或STAR找剪切位点。
介绍 nf-core / rnaseq是用于RNA测序数据的生物信息学分析管道。 该管道是使用构建的, 是一种工作流工具,可以以非常便携的方式跨多个计算基础架构运行任务。 它带有docker容器,使安装变得简单,结果可高度重现。...
首先进行预编译解压安装: 安装完成: 设置环境变量: 第二,源码安装: hisat2下载网址: 预编译安装同上,源码安装下载hisat2源码root权限下yum安装:暂时还未解决
主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!这里只是提供了各个分析流程的脚本,对于初学者来说是比较有好的。中提供了详细转录组上游分析...
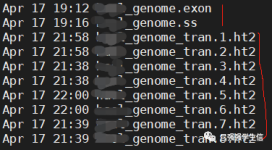
构建索引。
hisat2的用法 下载index文件 比对、排序、索引 质量控制 载入IGV,截图几个基因 hisat2的用法 本作业是比对到基因组,所以使用gapped or splices mapper,此流程已经更新。TopHat首次被发表已经是7年前,STAR的比对...

HISAT2 - StringTie - DESeq2 pipeline 进行bulk RNA-seq
HISAT2序列比对
标签: 生物信息学
HISAT2是一种快速、灵敏的比对程序,用于将下一代测序读数(全基因组、转录组和外显子组测序数据)与普通人群(以及单个参考基因组)进行比对。 1.建立索引 建立索引时间长,一般不需要自己建立,常见的基因组索引...
hisat2读取不到文件
标签: python
Warning: Could not open read file "SRR17972630_2" for reading;... Error: No input read files were valid (ERR): hisat2-align exited with value 1 我想要用hisat2文件将fastq对比为sam文件,结果报错说这样。
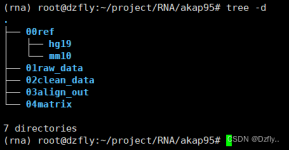
测试hisat2时遇到的问题
标签: linux
(ERR): Expected hisat2 to be in same directory with hisat-align: /home/vary2/src/hisat2-2.1.0/
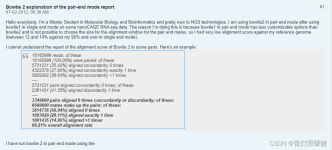
hisat2比对结果图文解析
标签: 大数据

索引(index)是帮助MySQL高效获取数据的数据结构(有效),在数据之外,数据库系统还维护着满足特定查找算法的数据结构,这些数据结构以某种方式引用(指向)数据, 这样就可以在这些数据结构上实现高级查找算法,...
Running HISAT2 Reporting The reporting mode governs how many alignments HISAT2 looks for, and how to report them. In general, when we say that a read has an alignment, we mean that it has a valid a...

bwa、bowtie2、tophat、hisat bwa bwa(Burrows-Wheeler Aligner) bwa文档说明 http://bio-bwa.sourceforge.net/bwa.shtml BWA用于将低差异的序列映射到一个大的参考基因组,如人类基因组。由BWA-ba...
推荐文章
- 企业数据管理数据备份与恢复_企业生产数据备份恢复方案-程序员宅基地
- 16. QML中的一些粒子特效_qml实现的特效-程序员宅基地
- Android开源项目及资源查速表_android tape queuefile-程序员宅基地
- 巨杉数据库 CTO 王涛:新一代分布式数据库-程序员宅基地
- 最全css居中:水平居中+垂直居中+水平/垂直居中总结_style 水平居中-程序员宅基地
- 【愚公系列】2024年02月 《网络安全应急管理与技术实践》 015-网络安全应急技术与实践(Web层-文件上传漏洞)-程序员宅基地
- 从数据仓库到大数据,数据平台这25年是怎样进化的?[转]-程序员宅基地
- 关于使用Java后台导入excel文件,读取数据后,更新数据库,并返回数据给到前端的相关问题总结_excel 导入时第一条导入后将第一条的数据返回-程序员宅基地
- 一例JAVA多线程访问卡死的现象_http-nio-8181-exec-4 线程过多导致卡死-程序员宅基地
- Linux调试器之gdb-程序员宅基地